
Anti-COL1A1 Antibody (CAB16891)
")
- SKU:
- CAB16891
- Product type:
- Antibody
- Reactivity:
- Human
- Reactivity:
- Mouse
- Reactivity:
- Rat
- Host Species:
- Rabbit
- Isotype:
- IgG
- Antibody Type:
- Polyclonal Antibody
- Research Area:
- Cell Biology
")
Description
| 抗体名: | Anti-COL1A1 Antibody |
| 抗体コード: | CAB16891 |
| 抗体サイズ: | 20uL, 50uL, 100uL |
| 申し込み: | WB IHC IF IP |
| 反応性: | Human, Mouse, Rat |
| 宿主種: | Rabbit |
| 免疫原: | A synthetic peptide of human COL1A1. |
| 申し込み: | WB IHC IF IP |
| 推奨希釈: | WB 1:500 - 1:2000 IHC 1:50 - 1:200 IF 1:50 - 1:200 IP 1:50 - 1:200 |
| 反応性: | Human, Mouse, Rat |
| ポジティブサンプル: | Mouse spleen, Mouse kidney, Rat lung |
| 免疫原: | A synthetic peptide of human COL1A1. |
| 精製方法: | Affinity purification |
| ストレージバッファ: | Store at -20'C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| アイソタイプ: | IgG |
| 順序: | Email for sequence |
| 遺伝子ID: | 1277 |
| Uniprot: | P02452 |
| セルラーロケーション: | Secreted, extracellular matrix, extracellular space |
| 計算された分子量: | 138kDa |
| 観察された分子量: | 139KDa |
| 同義語: | COL1A1, EDSC, OI1, OI2, OI3, OI4 |
| バックグラウンド: | This gene encodes the pro-alpha1 chains of type I collagen whose triple helix comprises two alpha1 chains and one alpha2 chain. Type I is a fibril-forming collagen found in most connective tissues and is abundant in bone, cornea, dermis and tendon. Mutations in this gene are associated with osteogenesis imperfecta types I-IV, Ehlers-Danlos syndrome type VIIA, Ehlers-Danlos syndrome Classical type, Caffey Disease and idiopathic osteoporosis. Reciprocal translocations between chromosomes 17 and 22, where this gene and the gene for platelet-derived growth factor beta are located, are associated with a particular type of skin tumor called dermatofibrosarcoma protuberans, resulting from unregulated expression of the growth factor. Two transcripts, resulting from the use of alternate polyadenylation signals, have been identified for this gene. |
| UniProt Protein Function: | COL1A1: Type I collagen is a member of group I collagen (fibrillar forming collagen). Defects in COL1A1 are the cause of Caffey disease (CAFFD); also known as infantile cortical hyperostosis. Caffey disease is characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age. Defects in COL1A1 are a cause of Ehlers-Danlos syndrome type 1 (EDS1); also known as Ehlers-Danlos syndrome gravis. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS1 is the severe form of classic Ehlers-Danlos syndrome. Defects in COL1A1 are the cause of Ehlers-Danlos syndrome type 7A (EDS7A); also known as autosomal dominant Ehlers-Danlos syndrome type VII. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS7A is marked by bilateral congenital hip dislocation, hyperlaxity of the joints, and recurrent partial dislocations. Defects in COL1A1 are a cause of osteogenesis imperfecta type 1 (OI1). A dominantly inherited connective tissue disorder characterized by bone fragility and blue sclerae. Osteogenesis imperfecta type 1 is non-deforming with normal height or mild short stature, and no dentinogenesis imperfecta. Defects in COL1A1 are a cause of osteogenesis imperfecta type 2 (OI2); also known as osteogenesis imperfecta congenita. A connective tissue disorder characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency. Defects in COL1A1 are a cause of osteogenesis imperfecta type 3 (OI3). A connective tissue disorder characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera, and dentinogenesis imperfecta. Defects in COL1A1 are a cause of osteogenesis imperfecta type 4 (OI4); also known as osteogenesis imperfecta with normal sclerae. A connective tissue disorder characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta. Genetic variations in COL1A1 are a cause of susceptibility to osteoporosis (OSTEOP); also known as involutional or senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mass, disruption of bone microarchitecture without alteration in the composition of bone. Osteoporotic bones are more at risk of fracture. A chromosomal aberration involving COL1A1 is found in dermatofibrosarcoma protuberans. Translocation t(17;22)(q22;q13) with PDGF. Belongs to the fibrillar collagen family. |
| UniProt Protein Details: | Protein type:Extracellular matrix; Secreted, signal peptide; Secreted Chromosomal Location of Human Ortholog: 17q21.33 Cellular Component: extracellular matrix; Golgi apparatus; extracellular space; endoplasmic reticulum lumen; collagen type I; extracellular region; secretory granule Molecular Function:identical protein binding; protein binding; platelet-derived growth factor binding; metal ion binding; extracellular matrix structural constituent Biological Process: response to peptide hormone stimulus; intramembranous ossification; extracellular matrix organization and biogenesis; response to cAMP; collagen fibril organization; positive regulation of transcription, DNA-dependent; embryonic skeletal development; response to estradiol stimulus; response to corticosteroid stimulus; extracellular matrix disassembly; protein transport; sensory perception of sound; visual perception; skeletal development; collagen biosynthetic process; endochondral ossification; response to drug; blood vessel development; receptor-mediated endocytosis; platelet activation; skin morphogenesis; osteoblast differentiation; collagen catabolic process; response to hyperoxia; response to hydrogen peroxide; blood coagulation; leukocyte migration; positive regulation of cell migration Disease: Osteogenesis Imperfecta, Type I; Ehlers-danlos Syndrome, Type Vii, Autosomal Dominant; Osteogenesis Imperfecta, Type Ii; Ehlers-danlos Syndrome, Type I; Osteogenesis Imperfecta, Type Iii; Osteoporosis; Caffey Disease; Osteogenesis Imperfecta, Type Iv |
| NCBI Summary: | This gene encodes the pro-alpha1 chains of type I collagen whose triple helix comprises two alpha1 chains and one alpha2 chain. Type I is a fibril-forming collagen found in most connective tissues and is abundant in bone, cornea, dermis and tendon. Mutations in this gene are associated with osteogenesis imperfecta types I-IV, Ehlers-Danlos syndrome type VIIA, Ehlers-Danlos syndrome Classical type, Caffey Disease and idiopathic osteoporosis. Reciprocal translocations between chromosomes 17 and 22, where this gene and the gene for platelet-derived growth factor beta are located, are associated with a particular type of skin tumor called dermatofibrosarcoma protuberans, resulting from unregulated expression of the growth factor. Two transcripts, resulting from the use of alternate polyadenylation signals, have been identified for this gene. [provided by R. Dalgleish, Feb 2008] |
| UniProt Code: | P02452 |
| NCBI GenInfo Identifier: | 296439504 |
| NCBI Gene ID: | 1277 |
| NCBI Accession: | P02452.5 |
| UniProt Related Accession: | P02452 |
| Molecular Weight: | |
| NCBI Full Name: | Collagen alpha-1(I) chain |
| NCBI Synonym Full Names: | collagen type I alpha 1 chain |
| NCBI Official Symbol: | COL1A1 |
| NCBI Official Synonym Symbols: | OI1; OI2; OI3; OI4; EDSC; EDSARTH1 |
| NCBI Protein Information: | collagen alpha-1(I) chain |
| UniProt Protein Name: | Collagen alpha-1(I) chain |
| UniProt Synonym Protein Names: | Alpha-1 type I collagen |
| UniProt Gene Name: | COL1A1 |
| UniProt Entry Name: | CO1A1_HUMAN |
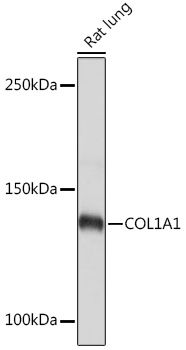 | Western blot analysis of extracts of Rat lung, using COL1A1 Rabbit pAb (CAB16891) at 1:500 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). Exposure time: 1s. |
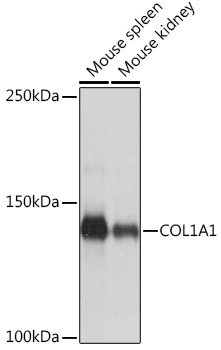 | Western blot analysis of extracts of various cell lines, using COL1A1 Rabbit pAb (CAB16891) at 1:500 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). Exposure time: 10s. |
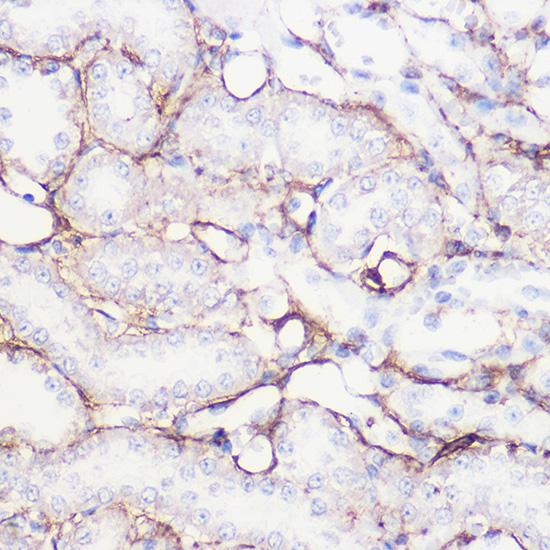 | Immunohistochemistry of paraffin-embedded rat kidney using COL1A1 Rabbit pAb (CAB16891) at dilution of 1:100 (40x lens). |
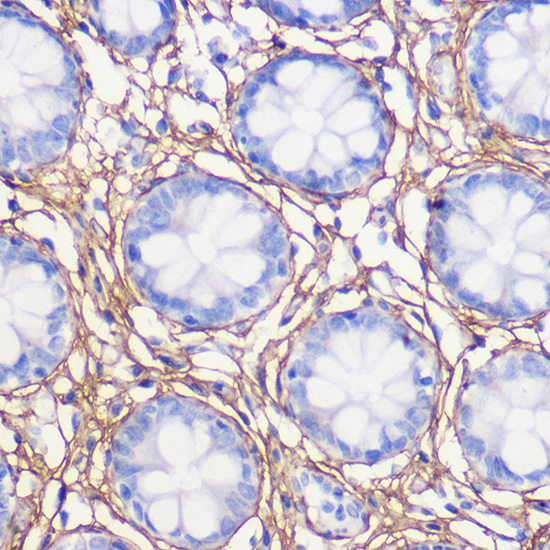 | Immunohistochemistry of paraffin-embedded human colon using COL1A1 Rabbit pAb (CAB16891) at dilution of 1:100 (40x lens). |
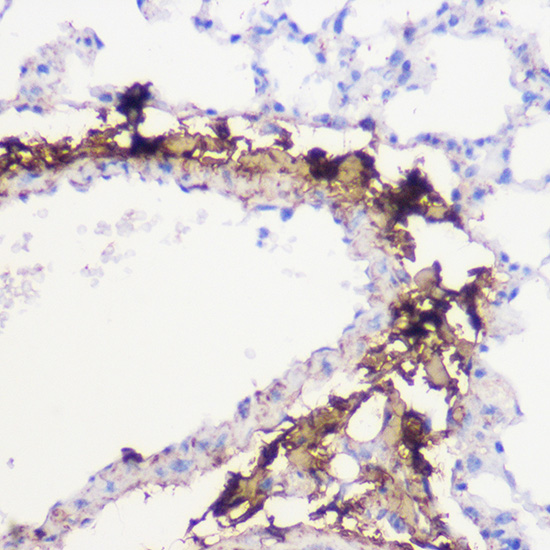 | Immunohistochemistry of paraffin-embedded mouse lung using COL1A1 Rabbit pAb (CAB16891) at dilution of 1:100 (40x lens). |