
Anti-C10orf2 Antibody (CAB5303)
")
- SKU:
- CAB5303
- Product type:
- Antibody
- Reactivity:
- Human
- Reactivity:
- Mouse
- Reactivity:
- Rat
- Host Species:
- Rabbit
- Isotype:
- IgG
- Research Area:
- Epigenetics and Nuclear Signaling
")
Frequently bought together:
Description
| 抗体名: | Anti-C10orf2 Antibody |
| 抗体コード: | CAB5303 |
| 抗体サイズ: | 20uL, 50uL, 100uL |
| 申し込み: | WB IF |
| 反応性: | Human, Mouse, Rat |
| 宿主種: | Rabbit |
| 免疫原: | Recombinant fusion protein containing a sequence corresponding to amino acids 385-684 of human C10orf2 (NP_068602.2). |
| 申し込み: | WB IF |
| 推奨希釈: | WB 1:500 - 1:2000 IF 1:50 - 1:200 |
| 反応性: | Human, Mouse, Rat |
| ポジティブサンプル: | MCF7 |
| 免疫原: | Recombinant fusion protein containing a sequence corresponding to amino acids 385-684 of human C10orf2 (NP_068602.2). |
| 精製方法: | Affinity purification |
| ストレージバッファ: | Store at -20'C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| アイソタイプ: | IgG |
| 順序: | EQAA GLRW SRFP DLNR ILKG HRKG ELTV FTGP TGSG KTTF ISEY ALDL CSQG VNTL WGSF EISN VRLA RVML TQFA EGRL EDQL DKYD HWAD RFED LPLY FMTF HGQQ SIRT VIDT MQHA VYVY DICH VIID NLQF MMGH EQLS TDRI AAQD YIIG VFRK FATD NNCH VTLV IHPR KEDD DKEL QTAS IFGS AKAS QEAD NVLI LQDR KLVT GPGK RYLQ VSKN RFDG DVGV FPLE FNKN SLTF SIPP KNKA RLKK IKDD TGPV AKKP SSGK KGAT TQNS EICS GQAP TPDQ PDTS KRSK |
| 遺伝子ID: | 56652 |
| Uniprot: | Q96RR1 |
| セルラーロケーション: | Mitochondrion matrix, mitochondrion nucleoid |
| 計算された分子量: | 60kDa/66kDa/77kDa |
| 観察された分子量: | 60kDa |
| 同義語: | TWNK, ATXN8, C10orf2, IOSCA, MTDPS7, PEO, PEO1, PEOA3, PRLTS5, SANDO, SCA8, TWINL |
| バックグラウンド: | This gene encodes a hexameric DNA helicase which unwinds short stretches of double-stranded DNA in the 5' to 3' direction and, along with mitochondrial single-stranded DNA binding protein and mtDNA polymerase gamma, is thought to play a key role in mtDNA replication. The protein localizes to the mitochondrial matrix and mitochondrial nucleoids. Mutations in this gene cause infantile onset spinocerebellar ataxia (IOSCA) and progressive external ophthalmoplegia (PEO) and are also associated with several mitochondrial depletion syndromes. Alternative splicing results in multiple transcript variants encoding distinct isoforms. |
| UniProt Protein Function: | PEO1: Involved in mitochondrial DNA (mtDNA) metabolism. Could function as an adenine nucleotide-dependent DNA helicase. Function inferred to be critical for lifetime maintenance of mtDNA integrity. In vitro, forms in combination with POLG, a processive replication machinery, which can use double-stranded DNA (dsDNA) as template to synthesize single-stranded DNA (ssDNA) molecules. May be a key regulator of mtDNA copy number in mammals. Defects in PEO1 are the cause of progressive external ophthalmoplegia with mitochondrial DNA deletions autosomal dominant type 3 (PEOA3). Progressive external ophthalmoplegia is characterized by progressive weakness of ocular muscles and levator muscle of the upper eyelid. In a minority of cases, it is associated with skeletal myopathy, which predominantly involves axial or proximal muscles and which causes abnormal fatigability and even permanent muscle weakness. Ragged- red fibers and atrophy are found on muscle biopsy. A large proportion of chronic ophthalmoplegias are associated with other symptoms, leading to a multisystemic pattern of this disease. Additional symptoms are variable, and may include cataracts, hearing loss, sensory axonal neuropathy, ataxia, depression, hypogonadism, and parkinsonism. Defects in PEO1 are a cause of sensory ataxic neuropathy dysarthria and ophthalmoparesis (SANDO). SANDO is a clinically heterogeneous systemic disorder with variable features resulting from mitochondrial dysfunction. It shares phenotypic characteristics with autosomal recessive progressive external ophthalmoplegia and mitochondrial neurogastrointestinal encephalopathy syndrome. The clinical triad of symptoms consists of sensory ataxic, neuropathy, dysarthria, and ophthalmoparesis. Defects in PEO1 are the cause of mitochondrial DNA depletion syndrome type 7 (MTDPS7); also known as spinocerebellar ataxia infantile-onset (IOSCA). A severe disease associated with mitochondrial dysfunction. Some patients are affected by progressive atrophy of the cerebellum, brain stem, the spinal cord, and sensory axonal neuropath. Clinical features include hypotonia, athetosis, ataxia, ophthalmoplegia, sensorineural hearing deficit, sensory axonal neuropathy, epileptic encephalopathy and female hypogonadism. Some individuals manifest a hepatocerebral phenotype characterized by liver insufficiency, increased serum and CSF lactate, hypotonia, psychomotor retardation and peripheral neuropathy. 3 isoforms of the human protein are produced by alternative splicing. |
| UniProt Protein Details: | Protein type:DNA replication; EC 3.6.4.12; Helicase; Mitochondrial Chromosomal Location of Human Ortholog: 10q24 Cellular Component: mitochondrial matrix Molecular Function:5'-3' DNA helicase activity; protease binding; single-stranded DNA binding Biological Process: mitochondrial DNA replication; mitochondrion organization and biogenesis; protein homooligomerization; transcription from mitochondrial promoter Disease: Mitochondrial Dna Depletion Syndrome 7 (hepatocerebral Type); Perrault Syndrome 5; Progressive External Ophthalmoplegia With Mitochondrial Dna Deletions, Autosomal Dominant, 3; Sensory Ataxic Neuropathy, Dysarthria, And Ophthalmoparesis |
| NCBI Summary: | This gene encodes a hexameric DNA helicase which unwinds short stretches of double-stranded DNA in the 5' to 3' direction and, along with mitochondrial single-stranded DNA binding protein and mtDNA polymerase gamma, is thought to play a key role in mtDNA replication. The protein localizes to the mitochondrial matrix and mitochondrial nucleoids. Mutations in this gene cause infantile onset spinocerebellar ataxia (IOSCA) and progressive external ophthalmoplegia (PEO) and are also associated with several mitochondrial depletion syndromes. Alternative splicing results in multiple transcript variants encoding distinct isoforms.[provided by RefSeq, Aug 2009] |
| UniProt Code: | Q96RR1 |
| NCBI GenInfo Identifier: | 74752111 |
| NCBI Gene ID: | 56652 |
| NCBI Accession: | Q96RR1.1 |
| UniProt Secondary Accession: | Q96RR1,Q6MZX2, Q6PJP5, Q96RR0, B2CQL2, |
| UniProt Related Accession: | Q96RR1 |
| Molecular Weight: | 60,400 Da |
| NCBI Full Name: | Twinkle protein, mitochondrial |
| NCBI Synonym Full Names: | chromosome 10 open reading frame 2 |
| NCBI Official Symbol: | C10orf2 |
| NCBI Official Synonym Symbols: | PEO; PEO1; SCA8; ATXN8; IOSCA; PEOA3; SANDO; TWINL; MTDPS7; PRLTS5 |
| NCBI Protein Information: | twinkle protein, mitochondrial |
| UniProt Protein Name: | Twinkle protein, mitochondrial |
| UniProt Synonym Protein Names: | Progressive external ophthalmoplegia 1 protein; T7 gp4-like protein with intramitochondrial nucleoid localization; T7-like mitochondrial DNA helicase |
| Protein Family: | Twinkle protein |
| UniProt Gene Name: | PEO1 |
| UniProt Entry Name: | PEO1_HUMAN |
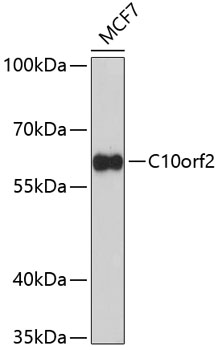 | Western blot analysis of extracts of MCF-7 cells, using C10orf2 Antibody (CAB5303) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Enhanced Kit (RM00021). Exposure time: 90s. |
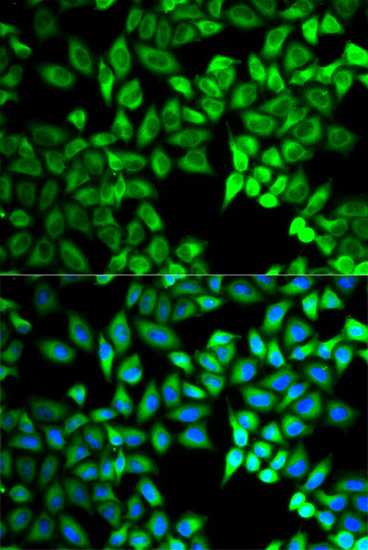 | Immunofluorescence analysis of A549 cells using C10orf2 antibody (CAB5303). Blue: DAPI for nuclear staining. |
View AllClose